Decoys are computer generated conformations of protein sequences that possess some characteristics of native proteins, but are not biologically real. The primary use of decoys is to test scoring, or energy, functions. All the decoys in the Decoys 'R' Us database can be downloaded.
The current version of the entire decoy set is available as a single tar and gzipped file. Subsets are also available, with the same directory organisation. Decoy sets can be downloaded from a special download area or by clicking on a decoy name in the hypertext version of this document.
The typographic convention used in this section is generally as follows:
Literal words which should be used exactly as written are represented
using a fixed width font (example: filename). Variable
names are represented using italics (example: filename).
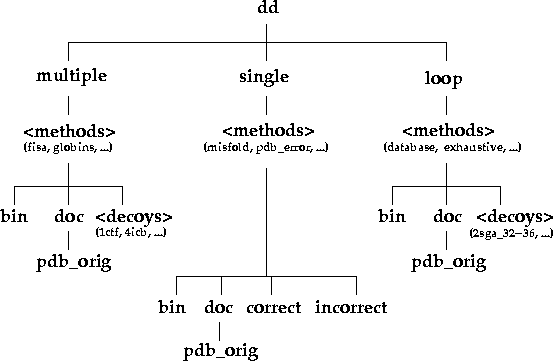
Under the top level directory (dd), there are three
directories: single, multiple, and
loop, which indicate the types of decoy sets present in
this database (Figure 1). single decoy sets are cases
where one one incorrect conformation is present for a given native
structure. multiple decoy sets are cases where a range of
conformations with different root mean square deviations (RMSD) to the
experimental structure are present. loop decoys contain
many conformations for a small stretch of sequence in the
protein. Each directory contains a file list which lists
the names of the decoy sets for each category. In the case of the
single decoy set the primary objective is to distinguish
the native conformation from the non-native one. In the case of the
multiple and loop decoy sets, the primary
objective is to select a conformation with a low RMSD to the
experimental one.
Figure 1. Directory organisation of the decoy database. The directories and their contents can be explored interactively via the www by clicking here.
All conformations are stored in Protein Data Bank (PDB) file
format [Bernstein et al, 1977].
Experimental conformations have generally been re-numbered to start
from 1, ignoring any chain breaks (the doc/pdb_orig
directory within a decoy set contains the original experimental
conformations). Multiple side chain conformations and hydrogen atom
positions have also been eliminated.
Any scoring function that requires the chain numbering to correspond exactly to the sequence (i.e., taking missing residues into account) should use the original experimental conformations. However, every effort has been made to collect decoys for experimental structures without chain breaks.
The PDB identifiers for the decoy sets are the same ones used by the creators of the decoys. In cases where the experimental structure has been superseded in the Protein Data Bank, the original names are used. A look up in the current version of the PDB <http://www.pdb.org> will automatically point to the superseded entry, if one exists.
The multiple decoy sets are listed in Table 1. Each decoy set is
in a directory with the same name (under the directory
dd/multiple, of course). Within that directory, each
protein has its own directory, which will contain all the decoy
conformations and the corresponding native conformation. Also
included will be a file called list, which simply lists
all the conformation (PDB) files in the directory and
rmsds, which gives the CA root mean square deviations
(cRMSDs; column 7) for each conformation (column 5). The mapping of
the list and rmsds files is currently 1:1,
but for extensibility purposes, only lines containing the string
cRMSD in the first column of the rmsds file
(grep ^cRMSD) should be used for this behaviour to be
guaranteed. In the future, all-atom and main chain RMSDs (denoted by
aRMSD and mRMSD respectively), may also be included in the
rmsds file. The other columns in the rmsds
file are reserved for future use. The RMSDs are calculated using the
program fit in the RAMP distribution <http://www.ram.org/computing/ramp/>,
a suite of programs to help in protein structure prediction.
Table 1. multiple decoy sets. To download a particular
set, click on the name of the set. Click
here to download all the multiple decoy sets.
| Name of set | Number of proteins | Average number of decoys per set (~) | Reference |
|---|---|---|---|
4state_reduced |
7 | 665 | [Park & Levitt, 1996] |
fisa |
6 | 1432 | [Simons et al, 1997] |
fisa_casp3 |
6 | 1432 | [Simons et al, 1997] |
hg_structal |
29 | 29 | [Samudrala et al, 1998c] |
ig_structal |
61 | 60 | [Samudrala et al, 1998c] |
ig_structal_hires |
20 | 19 | [Samudrala et al, 1998c] |
lattice_ssfit |
8 | 2000 | [Samudrala et al, 1999,Xia et al, 2000] |
lmds |
11 | 439 | [Keasar & Levitt, 1999] |
semfold |
6 | 12900 | [Samudrala & Levitt, 2002] |
vhp_mcmd |
1 | 6255 | [Fogolari et al, 2005] |
A directory name with the suffix of _u (for
unrefined) indicates that the decoy conformations for a given protein
have not been energy minimised, in cases where both minimised and
unminimised versions of a decoy are provided.
The bin directory (if it exists) contains shell
scripts or programs that will help manipulation of these decoys. The
doc directory contains any pertinent documentation. The
file NOTES (see example in Figure 2) contains details
about the decoy sets, including the primary source, the number of
decoys and the range of RMSDs for each protein, a short description,
any relevant comments, and references. For particular decoy sets, the
doc directory contains the original PDB files (as
available in the Protein Data Bank) under the pdb_orig
subdirectory.
Figure 2. Example of a NOTES file for the
multiple decoy sets. This particular
file is for the fisa decoy set.
----------------------------------------------------------------------- DESCRIPTION ----------------------------------------------------------------------- The fisa set contains decoys for four small alpha-helical proteins. The main chains for these decoys were generated using a fragment insertion simulated annealing procedure to assemble native-like structures from fragments of unrelated protein structures with similar local sequences using Bayesian scoring functions [Simons et al, 1997; PRIMARY SOURCE]. Side chains for these proteins were modelled with the software package SCWRL [Bower et al, 1997]. ----------------------------------------------------------------------- PRIMARY SOURCE ----------------------------------------------------------------------- Simons KT, Kooperberg C, Huang ES, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and bayesian scoring functions. J Mol Biol 268:209-225, 1997. ----------------------------------------------------------------------- SUMMARY ----------------------------------------------------------------------- Protein # cRMSD range Resolution/R Reference 1fc2 500 3.111 - 10.580 2.8/0.22 [Deisenhofer, 1981] 1hdd-C 500 2.769 - 12.915 2.8/0.24 [Kissinger et al, 1990] 2cro 500 4.288 - 12.599 2.4/0.20 [Mondragon et al, 1989] 4icb 500 4.754 - 14.130 1.6/0.19 [Svensson et al, 1992] average 500 3.731 - 12.556 2.4/0.21 ----------------------------------------------------------------------- COMMENTS ----------------------------------------------------------------------- All conformations were subjected to 500 steps of steepest descent minimusation using the CHARMM22b force-field [Brooks et al, 1983], ignoring electrostatic terms and using a cut-off of 12 A for non-bonded interactions. Resolution, R-factor, and Reference listed above are details about the experimental structure. ----------------------------------------------------------------------- REFERENCES ----------------------------------------------------------------------- Bower MJ, Cohen FE, Dunbrack RL. Prediction of protein side-chain rotamer from a backbone dependent rotamer library: a new homology modelling tool. J Mol Biol 267:1268-1282, 1997. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimisation, and dynamics calculations. J Comput Chem 4:187-217, 1994. Deisenhofer J. Crystallographic refinement and atomic models of a human fc fragment and its complex with fragment B of protein A from staphylococcus aureus at 2.9 and 2.8 angstroms resolution. Biochemistry 20:2361-2370, 1981. Kissinger CR, Liu BS, Martin-Blanco E, Kornberg TB, Pabo CO. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: a framework for understanding homeodomain-DNA interactions. Cell 63:579-590, 1990. Mondragon A, Wolberger C, Harrison SC. Structure of phage 434 cro protein at 2.35 angstroms resolution. J Mol Biol 205:179-188, 1989. Svensson LA, Thulin E, Forsen S. Proline cis-trans isomers in calbindin observed by x-ray crystallography. J Mol Biol 223:601-606, 1992. -----------------------------------------------------------------------
The single decoy sets are listed in Table 2. There
is a directory for each single decoy set under
dd/single. Within that directory are two directories:
correct and incorrect, which respectively
contain the correct and the corresponding incorrect conformations for
a given protein. Also included in both directories is a file called
list which lists the conformation (PDB) files in those
directories. The mapping of the list files in the
correct and incorrect directories is 1:1,
even if there are fewer correct conformations than incorrect
conformations.
Table 2. single decoy sets. To download a particular
set, click on the name of the set. Click
here to download all the single decoy sets.
| Name of set | Number of proteins/decoys | Reference |
|---|---|---|
misfold |
26 | [Holm & Sander, 1992] |
pdb_error |
3 | [Branden & Jones, 1990] |
As with the multiple decoy sets, doc and
bin serve to provide additional documentation and
executables to make processing of a given set easier. Also the
doc/pdb_orig directory contains the original experimental
structures. The format of the NOTES file is slightly
different than that used for the multiple decoy sets (see
example in Figure 3). Here, the RMSD ranges are omitted, but a summary
line is provided for each correct and incorrect decoy
conformation.
Figure 3. Example of a NOTES file for
single decoy sets. This particular file is for the
pdb_error decoy set.
----------------------------------------------------------------------- DESCRIPTION ----------------------------------------------------------------------- The pdb_error set contains coordinates for pairs of experimental structures in cases where one of the pair has been substantially refined or found to contain errors (this structure is designated as incorrect). ----------------------------------------------------------------------- PRIMARY SOURCE ----------------------------------------------------------------------- There is no primary source as both the correct and incorrect structures are directly obtained from the PDB and are produced by different sources. However, a general source to cite would be [Branden & Jones, 1990]. ----------------------------------------------------------------------- SUMMARY ----------------------------------------------------------------------- Correct Incorrect Resolution/R Reference 2f19 1f19 2.8/0.18 [Lascombe et al, 1992] 3hfl 2hfl 2.6/0.29 [Cohen et al, 1995] 5fd1 2fd1 1.9/0.21 [Stout, 1993] 5rxn 5rxnon1fdx 1.2/0.14 [Watenpaugh, 1984] ----------------------------------------------------------------------- COMMENTS ----------------------------------------------------------------------- Resolution, R-factor, and Reference listed above are details about the experimental (correct) structure. The incorrect structures are no longer present in the PDB. ----------------------------------------------------------------------- REFERENCES ----------------------------------------------------------------------- Branden CI, Jones TA. Between objectivity and subjectivity. Nature 343:687-689, 1990. Cohen GH, Sheriff S, Davies DR. The refined structure of the monoclonal antibody hy(slash)hel-5 with its antigen hen egg white lysozyme. To be published, 1995. Lascombe MB, Alzari PM, Poljak RJ, Nisonoff A. Three-dimensional structure of two crystal forms of fab r19.9, from a monoclonal anti-arsonate antibody. Proc Natl Acad Sci USA 89:9429-9433, 1992. Stout CD. Crystal structures of oxidized and reduced azotobacter vinelandii ferredoxin at ph 8 and ph 6. J Biol Chem 268:25920-25927, 1993. -----------------------------------------------------------------------
Table 3 lists the loop decoy sets. The directory
name for each loop decoy set, under
dd/loop/method/loop-set,
takes on the form
protein_start-stop.
start and stop signify the residue ranges for a
given loop that varies in conformation, for example
3dfr_20-40. All the loop conformations are stored in a
single file, with the name
protein_start-stop.loops.pdb.
Within the directory for each loop set, the file
loop_data contains information about the loop, containing
the range of residues, the name of the experimental structure with the
right orientation (so the loops can just be inserted into the
structure), the name of the file containing all the loops, and the
number of lines per loop in that file (see example in Figure 4).
Table 2. loop decoy sets. To download a particular
set, click on the name of the set. Click
here to download all the single decoy sets.
| Name of set | Number of sets | Average number of loops per set (~) | Reference |
|---|---|---|---|
abm_database |
4 | 200 | [Samudrala & Moult, 1998b] |
Figure 4. Example of a loop_data file for
loop decoy sets. This particular file is for the
1vfa_205-212 decoy set.
205 212 1vfa.pdb 1vfa_205-212.loops.pdb 78
In all the loop sets, there is only one stretch of
sequence that varies---the rest of the protein is held constant. If
more than one stretch varies, then the set is considered to belong in
the multiple decoy set. The bin,
doc, and doc/pdb_orig directories serve the
the same purpose as in the multiple and
single decoy set. The format of the NOTES
file is similar to the format used for multiple decoy
sets, but the summary line information contains details about size and
range of the loop residues, the sequence, the number of loop
conformations, and the CA RMSD ranges (see example in Figure 5).
Figure 5. Example of a NOTES file for
loop decoy sets. This particular file is for the
abm_database set.
-----------------------------------------------------------------------
DESCRIPTION
-----------------------------------------------------------------------
The abm_database set contains loop conformations for the d1.3 antibody
(1vfa). The main chains for these loops were generated using a
database procedure [Pedersen et al, 1992]. Side chains were
constructed using the program scgen [Samudrala & Moult, 1998a]. These
loops were generated to test the ability of a graph theoretical clique
finding method to select the best set of loop conformations taking the
environment context into account [Samudrala & Moult, 1998; PRIMARY
SOURCE].
-----------------------------------------------------------------------
PRIMARY SOURCE
-----------------------------------------------------------------------
Samudrala R, Moult J.
A graph-theoretic algorithm for comparative modelling of protein structure.
J Mol Biol 279:287-302, 1998.
-----------------------------------------------------------------------
SUMMARY
-----------------------------------------------------------------------
Loop Size Sequence # cRMSD range Resolution/R Reference
1vfa_47-55 9 LVYYTTTLA 176 0.663 - 5.287 1.8/0.15 [Bhat et al, 1994]
1vfa_90-97 8 HFWSTPRT 166 0.646 - 5.247 1.8/0.15 [Bhat et al, 1994]
1vfa_158-166 9 MIWGDGNTD 168 0.402 - 6.239 1.8/0.15 [Bhat et al, 1994]
1vfa_205-212 8 RERDYRLD 216 0.458 - 5.435 1.8/0.15 [Bhat et al, 1994]
average 9 182 0.542 - 5.552 1.8/0.15
-----------------------------------------------------------------------
COMMENTS
-----------------------------------------------------------------------
Resolution, R-factor, and Reference listed above are details about the
experimental structure.
-----------------------------------------------------------------------
REFERENCES
-----------------------------------------------------------------------
Bhat TN, Bentley GA, Boulot G, Green MI, Tello D, Dall'acqua W, Souchon H, Schwarz FP, Mariuzza RA, Poljak RJ.
Bound water molecules and conformational stabilization help mediate an antigen-antibody association.
Proc Nat Acad Sci USA 91:1089-1093, 1994.
Pedersen J, Searle S, Henry A, Rees AR.
Antibody modelling: Beyond homology.
Immunomethods 1:126-136, 1992.
Samudrala R, Moult J.
Determinants of side chain conformational preferences in protein structures
Protein Eng, 1998 (in press).
-----------------------------------------------------------------------
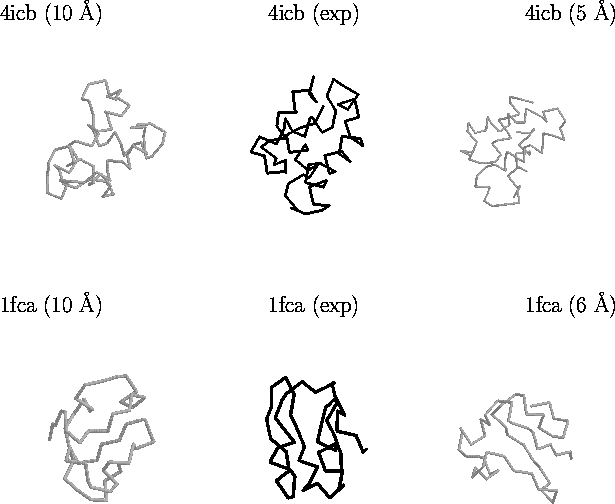
As mentioned above, the goal of this endeavour is to collect
decoys which scoring functions cannot distinguish from the native
conformation. Figure 6 illustrates this for two proteins in the
lattice_ssfit
decoy set.
Figure 6. Example of decoys in the lattice_ssfit
decoy set. Shown are decoys for two proteins: Calbindin (PDB code
4icb) which is an alpha-helical protein, and Ferrodoxin (PDB code
1fca). The structures on the far left represent grossly incorrect
structures with a good score by different scoring functions.
Structures in the middle are the experimental conformations.
Structures on the far right are selections by an all-atom scoring
function [Samudrala & Moult, 1998a]. All
structures depicted above are compact and have the native secondary
structure.
The maintainer has taken great pains to ensure that the people who have submitted decoys have given their permission and have been properly attributed. In any effort of this size, there are bound to be mistakes. If you spot a mistake, please let me know so I can fix it as soon as possible.
If you find any of the decoys useful and use it in a published work, please give credit where due. Also, please realise that all the contributors for this database have readily and openly published their work. People who use these database and publish their results equally openly would be making the best use of this effort.
If you wish to contribute decoy sets, please contact dd@compbio.org.
A special thank you goes out to the experimental community who have made all their experimental data publicly available. However, the efforts of many people make this database possible:
4state set)
lmds set)
misfold set)
fisa sets)
globins and 4state sets; general help)
fisa sets)
misfold set)
globins, immunoglobulins, and 4state sets; general help)
lattice_ssfit set)